A most sophisticated stowaway
19 Nov 2015 by Evoluted New Media
Unravelling Helicobacter pylori’s gut-grabbing grip has revealed some truly unique evolutionary adaptations – including a remarkable ability to attach to gastric epithelium








Unravelling Helicobacter pylori’s gut-grabbing grip has revealed some truly unique evolutionary adaptations – including a remarkable ability to attach to gastric epithelium
If it weren’t for the huge disease burden it carries, one couldn’t help but admire the resilience of the small spiral-shaped Gram-negative bacterium Helicobacter pylori. It is able to survive and thrive in the most hostile environment in the human body, the stomach.
This is an environment that has not only evolved to degrade and digest its food contents through a combination of proteases and high acidity, but also to kill bacteria and other potential pathogens ingested with meals. Remarkably, H. pylori infection, unless treated, can be life-long. The bacterium is also exceedingly successful at the population level, with more than one-half of the global population affected. Furthermore, it is pathogenic. H. pylori infection can lead to peptic ulceration and it is the strongest known risk factor for gastric cancer. In fact, H. pylori is the first bacterium to be classified as a carcinogen by the World Health Organization's International Agency for Research on Cancer.
The key to H. pylori’s successful pathogenicity lies in the unique evolutionary adaptations it uses to colonise the stomach. It is believed that these mechanisms have co-evolved with humans over millennia, as the existence of the bacterium can be traced back to 58,000 years ago, and beyond1.
What then are these adaptations that allow the bacterium to survive in such a hostile environment? One unique adaptation of H. pylori to life in the host’s stomach is its ability to counteract gastric acid by producing ammonia from urea. This is the result of the activity of a bacterial urease enzyme, and in fact is the basis of a widespread diagnostic test for H. pylori infection – the urease breath test (UBT).
[caption id="attachment_50365" align="aligncenter" width="620"]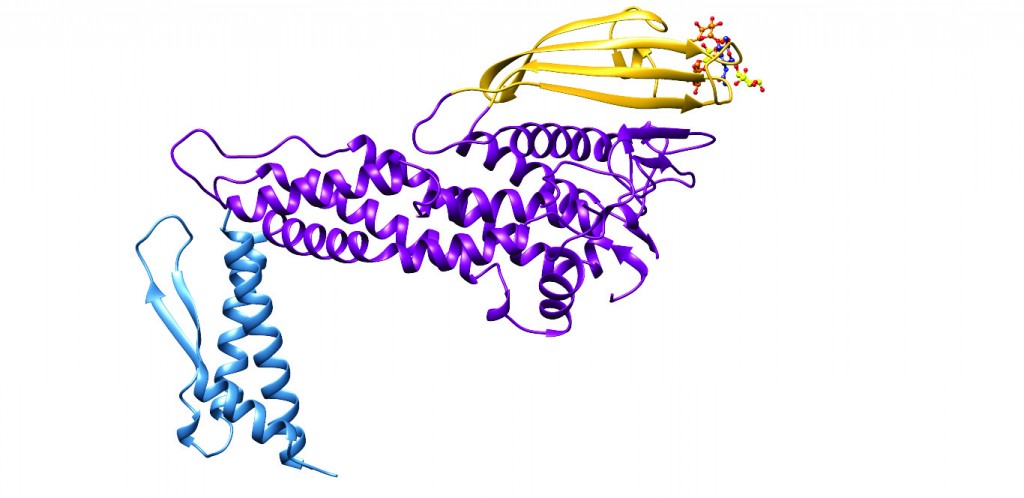 Figure 1: Ribbon diagram of the three-dimensional structure of the extracellular domain of the H. pylori adhesin BabA obtained through X-ray crystallography6. Three major regions can be recognised, the handle (blue), the head (violet) and the crown (gold). Located near the tip of the (golf putter-shaped) adhesin, is the shallow site responsible for binding of Lewisb.[/caption]
Figure 1: Ribbon diagram of the three-dimensional structure of the extracellular domain of the H. pylori adhesin BabA obtained through X-ray crystallography6. Three major regions can be recognised, the handle (blue), the head (violet) and the crown (gold). Located near the tip of the (golf putter-shaped) adhesin, is the shallow site responsible for binding of Lewisb.[/caption]
But neutralising the gastric acid as the sole strategy is not enough to ensure long-term survival. In addition to its acid-neutralising properties, H. pylori is a highly motile microorganism. It migrates away from the highly acidic lumen of the stomach to hide underneath the protective gastric mucus layer, where the pH is close to neutral, due to the constant release of bicarbonate by the stomach epithelium. How does the pathogen find its way in the labyrinth of mucus channels? Recent work has shown that H. pylori uses a bacterial chemoreceptor called TlpB for sensing of tissue-derived urea, to locate the surface of the epithelium2,3.
A series of pathogenicity factors are also important, such as the cytotoxin associated gene (cagA) and vacuolating cytotoxin A (VacA) as well as a series of molecules thought to play a role in adhesion to the epithelium. Interestingly, these genes are absent from the genome of the related bacterium Helicobacter hepaticus4, which does not colonise the stomach, but is similarly suspected to cause chronic hepatic inflammation and cancer.
One of its main survival mechanisms, without which infection and virulence significantly decreases, is the use of its adhesion protein BabA to attach to Lewisb antigens – sugars naturally expressed on healthy gastric epithelium. By being able to attach directly to epithelial cells, the bacteria evade clearance from the stomach during mucus turnover and local colonisation is sustained. While BabA (from blood group antigen-binding adhesin) was first discovered in 19985, the molecular mechanism through which it attaches to Lewisb antigens had remained elusive until now.
Using X-ray crystallography, the molecular basis of Lewisb antigen binding by BabA was revealed in unprecedented atomic detail. Published in Science Advances6, this showed for the first time that BabA uses a unique groove at its tip to bind Lewisb (Figure 1). The binding interaction is mediated by a network of hydrogen bonds that result in the highly specific recognition of Lewisb, but not other closely related sugars. Successful binding is completely dependent on the integrity of the network – if a few of these interactions are disrupted, BabA completely loses affinity for Lewisb.
[caption id="attachment_50366" align="aligncenter" width="620"]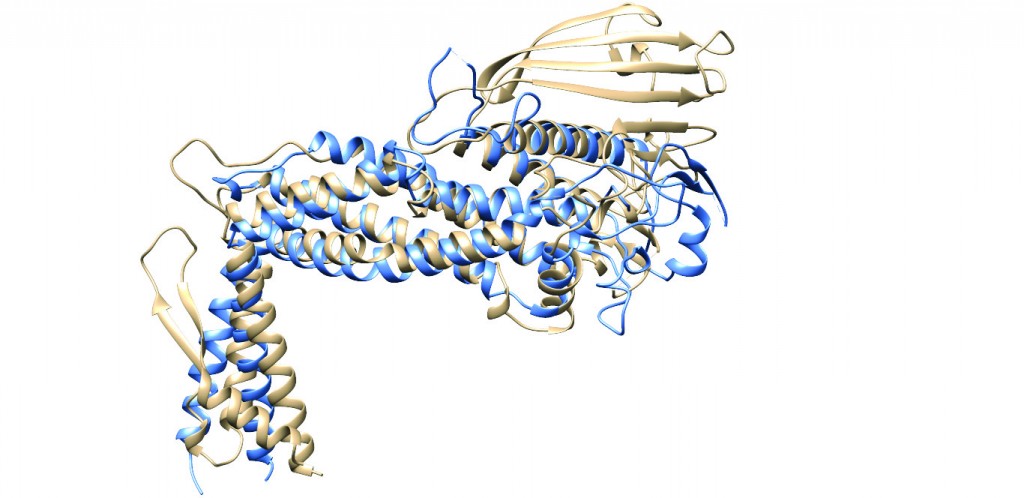 Figure 2: Ribbon diagrams representing the superimposed structures of the extracellular regions of BabA (tan)6 and SabA (blue)7, showing the similarity in the general fold between the two H. pylori adhesins. This finding was quite unexpected due to the low level of sequence identity (26%) between the extracellular regions of the two proteins at the amino acid level. Note the same marked kinked angle between the handle and the head region, and the lack of a crown region in SabA.[/caption]
Figure 2: Ribbon diagrams representing the superimposed structures of the extracellular regions of BabA (tan)6 and SabA (blue)7, showing the similarity in the general fold between the two H. pylori adhesins. This finding was quite unexpected due to the low level of sequence identity (26%) between the extracellular regions of the two proteins at the amino acid level. Note the same marked kinked angle between the handle and the head region, and the lack of a crown region in SabA.[/caption]
When analysing the structure of BabA, we were surprised by four main findings: First, the marked kinked angle between the handle and the head region – a feature which appears to be shared with the only other H. pylori adhesin for which a structure is known, called SabA7 (Figure 2). Secondly, the existence of a putatively novel carbohydrate recognition domain, represented by a beta-sheet region, which due to its location above the head region, we decided to call the ‘crown’ region (Figure 1). We have not been able to find any related sequences in other genes or organisms, suggesting that the crown could be a distinct recognition domain that is a specific adaptation of H. pylori.
Thirdly, we were surprised to find that most variations to the BabA amino acid sequence between various H. pylori strains mapped to a very short stretch of amino acids located within the Lewisb binding site (Figure 3). We termed this region the ‘hypervariable crown loop’ (HCL). In fact, due to its involvement in ligand binding, we believe the HCL to guard some secrets, which could in the future explain the marked differences in Lewisb binding affinity and specificity observed between H. pylori strains.
Finally, we were initially surprised by the low binding affinity of BabA for Lewisb (KD = 252 ?M), as determined through isothermal titration calorimetry. However such a low binding affinity is well in line with the known carbohydrate affinities of other lectins and the shallow nature of the binding pocket of BabA. H. pylori appears to compensate for such low binding affinity by presenting a multitude of adhesins on its surface, which in turn interacts with a multitude of carbohydrate ligands on the surface of the epithelium. This principle is known as avidity and is not dissimilar from the principle underlying the tight binding of Velcro strips.
[caption id="attachment_50367" align="aligncenter" width="620"]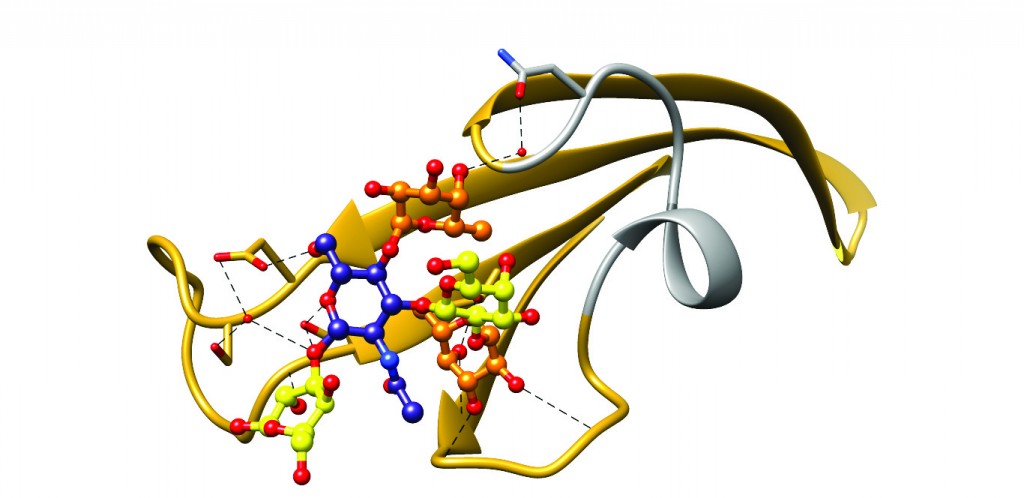 Figure 3: Ribbon diagram of the BabA crown region complexed with its physiological ligand Lewisb. Hydrogen bonds responsible for binding are depicted using black dashed lines. The short stretch corresponding to the hypervariable crown loop is shown in grey. This region is highly variable between strains and contains only one of the eight amino acids identified to mediate Lewisb binding in BabA J99 (N206, shown near the top of the HCL)6.[/caption]
Figure 3: Ribbon diagram of the BabA crown region complexed with its physiological ligand Lewisb. Hydrogen bonds responsible for binding are depicted using black dashed lines. The short stretch corresponding to the hypervariable crown loop is shown in grey. This region is highly variable between strains and contains only one of the eight amino acids identified to mediate Lewisb binding in BabA J99 (N206, shown near the top of the HCL)6.[/caption]
Is there any translational value to the elucidation of the carbohydrate binding site in BabA? To answer this question, we need to briefly look at the therapeutic options available for treatment of H. pylori infection. There is no vaccination currently, but there are various treatments for eradication of H. pylori. The standard treatment is a triple therapy, which consists of a combination of antibiotics (e.g. Amoxicillin and Clarithromycin, or Metronidazole), and an oral proton pump inhibitor (e.g. omeprazole), or a quadruple therapy (with or without bismuth) in more difficult cases. However the triple therapy achieves only about 70% cure rate8. The main reasons are issues with compliance (the therapy usually last for 2 weeks) and an increasing background of resistance to the antibiotics used (in particular to Clarithromycin)8. None of the available treatment options can achieve 100% eradication of the infection. Perhaps the cure rate could be increased by additionally interfering with important survival mechanisms of H. pylori, such as the epithelial attachment and gastric acid evasion strategy. For example, this may be achievable by antagonising the adhesive properties of BabA and other adhesins.
Indeed, there have been attempts to inhibit attachment of H. pylori, for example by using mucilaginous watery extracts of edible plants such as okra (Abelmoschus esculentus)9, a plant traditionally used for the treatment of gastritis, or from other culinary or medical plants10. The elucidation of the BabA binding site will enable a more rational, drug design-based approach in the search for efficient anti-adhesive compounds, of the type favoured by the pharmaceutical industry.
Taken together, we have for the first time shed a detailed light on the molecular basis of the intricate mechanisms underlying the adhesion of H. pylori to the gastric epithelium via the adhesin BabA, enabling the bacterium to bind tightly to a less hostile tissue niche. This may pave the way for new pharmacological treatment strategies.
References:
1. Linz B, Balloux F, Moodley Y, Manica A, Liu H, Roumagnac P, et al. An African origin for the intimate association between humans and Helicobacter pylori. Nature. 2007;445: 915–8. doi:10.1038/nature05562. 2. Croxen MA, Sisson G, Melano R, Hoffman PS. The Helicobacter pylori chemotaxis receptor TlpB (HP0103) is required for pH taxis and for colonization of the gastric mucosa. J Bacteriol. 2006;188: 2656–65. doi:10.1128/JB.188.7.2656-2665.2006. 3. Huang JY, Sweeney EG, Sigal M, Zhang HC, Remington SJ, Cantrell MA, et al. Chemodetection and Destruction of Host Urea Allows Helicobacter pylori to Locate the Epithelium. Cell Host Microbe. 2015;18: 147–56. doi:10.1016/j.chom.2015.07.002. 4. Suerbaum S, Josenhans C, Sterzenbach T, Drescher B, Brandt P, Bell M, et al. The complete genome sequence of the carcinogenic bacterium Helicobacter hepaticus. Proc Natl Acad Sci U S A. 2003;100: 7901–6. doi:10.1073/pnas.1332093100. 5. Ilver D, Arnqvist A, Ogren J, Frick IM, Kersulyte D, Incecik ET, et al. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science. 1998;279: 373–7. 6. Hage N, Howard T, Phillips C, Brassington C, Overman R, Debreczeni J, et al. Structural basis of Lewisb antigen binding by the Helicobacter pylori adhesin BabA. Sci Adv. 2015;1: 1–9. 7. Pang SS, Nguyen STS, Perry AJ, Day CJ, Panjikar S, Tiralongo J, et al. The three-dimensional structure of the extracellular adhesion domain of the sialic acid-binding adhesin SabA from Helicobacter pylori. J Biol Chem. 2014;289: 6332–40. doi:10.1074/jbc.M113.513135 8. Malfertheiner P, Megraud F, O’Morain CA, Atherton J, Axon ATR, Bazzoli F, et al. Management of Helicobacter pylori infection--the Maastricht IV/ Florence Consensus Report. Gut. 2012;61: 646–64. doi:10.1136/gutjnl-2012-302084. 9. Messing J, Thöle C, Niehues M, Shevtsova A, Glocker E, Borén T, et al. Antiadhesive properties of Abelmoschus esculentus (Okra) immature fruit extract against Helicobacter pylori adhesion. PLoS One. 2014;9: e84836. doi:10.1371/journal.pone.0084836. 10. O’Mahony R, Al-Khtheeri H, Weerasekera D, Fernando N, Vaira D, Holton J, et al. Bactericidal and anti-adhesive properties of culinary and medicinal plants against Helicobacter pylori. World J Gastroenterol. 2005;11: 7499–507. Available: http://www.ncbi.nlm.nih.gov/pubmed/16437723.
Acknowledgements:
This article is based on a research study conducted by the following researchers at the University of Nottingham and AstraZeneca R&D: N. Hage, T. Howard, C. Phillips, C. Brassington, R. Overman, J. Debreczeni, P. Gellert, S. Stolnik, G. S. Winkler and F. H. Falcone. The authors would also like to thank G. Davies and G. Holdgate of AstraZeneca R&D for useful discussions and assistance in collecting ITC data and the staff of ESRF (Grenoble, France) and DLS (Harwell, UK), in particular G. Leonard, for assistance in collecting X-ray crystallography data. This research was funded by the Engineering and Physical Sciences Research Council (EPSRC; grant EP/I01375X/1) and AstraZeneca R&D.
The authors:
Dr Franco H. Falcone is an Associate Professor at the School of Pharmacy, University of Nottingham.
Mr Naim Hage is a registered Pharmacist and a PhD student at the School of Pharmacy, University of Nottingham.
