Improving your protein predictions
Measuring zeta potential is a valuable way to predict how a protein will behave in solution - but there are problems with the method. Here we learn of a new way to improve accuracy
Measuring zeta potential is a valuable way to predict how a protein will behave in solution - but there are problems with the method. Here we learn of a new way to improve accuracy
Developing protein-based products demands an understanding of how they behave in solution since this underpins our ability to predict aggregation behaviour and to determine appropriate conditions for storage, crystallisation, and other processing parameters. Zeta potential, which is related to surface charge, is one of the parameters used in predicting the stability of protein-based preparations, but denaturation of the sample during measurement has always posed a particular challenge. The tendency for aggregation to occur at the measurement electrodes leads to measurement of denatured protein, which gives a result that is markedly different from the zeta potential of the native protein.
There is a new method which improves accuracy and repeatability when measuring the zeta potential of proteins – separating the measured molecules from the electrodes, using a diffusion barrier, means that false readings caused by aggregates of the sample can be avoided. Results comparing different methods, measured using a Zetasizer Nano demonstrates both the underlying issue and the advantage of applying this new method.
Proteins fold into pseudo stable structures that are sensitive to environmental conditions such as temperature, pH, and both the type and concentration of salt present. Stability of a protein’s tertiary structure can be achieved through a number of energy minimising mechanisms, including:
• incorporation of hydrophobic residues (amino acids) within the folded protein
• formation of disulphide cross links, which reduce entropic contributions
• Hydrogen bonding with neighbouring hydrophilic residues.
Unfortunately, all three of these stabilising mechanisms can be disrupted when proteins interact with charged metal electrodes during a measurement process. Protein adsorption, redox reactions at the disulphide bond, as well as deamidation and oxidation of surface side chain ammonium groups and histidine residues respectively, can all lead to protein denaturation.
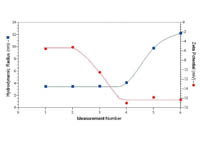 |
| Figure 1: Size and zeta potential results for human serum albumin in PBS for sequential electrophoretic light scattering measurements |
A common symptom of protein denaturation during the course of a zeta potential measurement using electrophoretic light scattering (ELS) is a time-dependent increase in hydrodynamic size, accompanied by a reduction in the measured zeta potential to more negative values.
Consider for example the results shown in Figure 1 for human serum albumin in phosphate buffered saline (PBS), where the hydrodynamic size was measured using dynamic light scattering (DLS) just prior to each zeta potential measurement using ELS. An increase in size is first noted in measurement number 4, indicating that the third zeta potential measurement induced a change in the size of the protein. Concurrent with the increase in size is a decrease in the measured zeta potential.
If the aggregation were non-denaturing, no significant change in zeta potential would be expected, as it is a function of the surface charge density rather than the total charge. The concurrent reduction in zeta potential with aggregation therefore suggests the likelihood of a change in the protein’s tertiary structure as a result of sample denaturation.
A number of potential solutions to the ELS protein denaturation problem have previously been considered. These include:
• simultaneously monitoring the size and stopping the measurements when a change is observed
• using only short bursts of applied voltage to minimise the “rate” of denaturation
• integrating multiple detectors, to reduce the total time that the field is applied by attempting to improve signal averaging.
However, while these approaches should improve results, they do not get to the heart of the matter. If the interactions of the protein at the charged metal electrodes could be reduced or even eliminated, then the need to minimise the consequences of protein aggregation would be neatly removed at source.
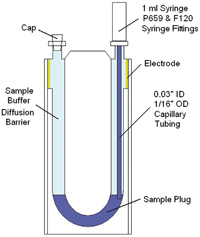 |
| Figure 2: Schematic of a disposable ZP cell, illustrating the use of a narrow needle to inject a small (< 50 ?l) plug of sample into a buffer filled cell |
Termed the ‘plug injection’ or ‘diffusion barrier’ technique by the scientists at Malvern Instruments who developed it, this new approach does just that. The method eliminates protein denaturation during zeta potential measurements by isolating a small sample volume (< 50 ?L) from the electrodes, in a fashion similar to that used in Capillary Electrophoresis (CE) instrumentation, where the sample is injected into a buffer stream.
Ideally, the sample should be monodisperse, i.e. contain no aggregates. The size distribution should consist of a single peak and the polydispersity index (PDI), or percent polydispersity, should be as low as possible. An increase in the PDI after a measurement is an excellent indicator that aggregates have begun to form.
Figure 2 shows a schematic of the disposable capillary zeta potential cell for the Zetasizer Nano system, illustrating the components needed and approach used to inject a small (< 50 ?L) plug into the measurement zone of a buffer filled cell.
Having rinsed the disposable capillary cell with ethanol, followed by water and sample buffer, all that is needed is to fill the folded capillary cell with the buffer in which the protein is dissolved using the usual Luer fitting syringe. It is then helpful if the cell is put into the Zetasizer Nano for 2-3 minutes to allow the temperature to equilibrate and reduce any temperature-related fluid motion such as convection.
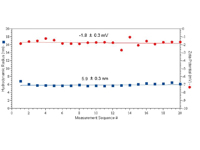 |
| Figure 3: Zeta potential and hydrodynamic diameter results for 10 mg/ml IgG in PBS, measured using the diffusion barrier approach |
The cell should then be removed and between 20 and 100 µL of the sample pipetted directly into the bottom of the U bend where the mobility measurement is made. This is best achieved using a gel- loading tip.
In order to produce optimum data, any mixing of the sample with the buffer should be kept to a minimum during loading. If the cell is kept upright and handled carefully, the sample will not diffuse around the cell. It is also important to minimise density and temperature gradients across the buffer/sample interface and ensure the sample remains within the measurement zone for more than enough time to complete the zeta potential measurements. To do this the sample must be isolated from the electrodes using the buffer in which the sample is prepared, and the temperature of both barrier and sample should be equal. Under normal temperatures and diffusion rates the amount of time needed for the sample to diffuse to the electrodes is measured in hours.
Using the method described above, this diffusion barrier facilitates multiple zeta potential measurements of low volume samples, avoiding interactions with the electrodes.
In principle, making a zeta potential measurement using the diffusion barrier technique is exactly the same as making a conventional measurement using the Zetasizer Nano. Light scattered by a molecule or particle undergoing electrophoresis is combined with a reference beam and the rate of change of phase of the combined beam is measured and used to calculate the electrophoretic mobility, and thus the zeta potential. It is recommended that a size measurement of the sample is made before and after the zeta potential measurement
In practise, at the higher buffer conductivities typical of physiological saline solutions, it is helpful to reduce the applied voltage and have a rest phase between repeat measurements to limit Joule heating.
A typical set of results for protein zeta potential measurements collected using the diffusion barrier approach with the Zetasizer Nano automatic measurement mode is shown in Figure 3. A 50 ?L sample of 10 mg/mL IgG in PBS (pH 7.4 & I = 130 mM) was measured. As indicated, both the hydrodynamic size and zeta potential are constant across 20 sequential size and zeta potential measurement sets.
These results provide clear evidence that the tertiary structure of the protein does not vary across the time span of the experiment, resulting in a marked improvement in the repeatability of zeta potential data.
By applying this new diffusion barrier method it is possible to eliminate aggregation effects caused by the interaction of protein samples and electrodes during zeta potential measurements using Electrophoretic Light Scattering. By minimising sample volume and separating the protein from the electrodes, the new method facilitates a simple, robust solution that enables protein scientists to deliver more accurate, repeatable zeta potential results.
Authors: Hanna Jankevics, Technical specialist, biophysical characterisation, & Malcolm Connah, Product Marketing Manager, Malvern Instruments
Contact
t: +44 (0)1684 892456
f:+ 44 (0)1684 892789
w: www.malvern.com









