The necrosome and the killer zombie proteins
28 Jan 2019 by Evoluted New Media






As cell biologists gets to grips with the relatively recent discovery of non-apoptotic programmed cell death and the fact that its dysfunction can lead to disease, Dr Emma Petrie takes us through the proteomics of the necrosome.
As we walk around in our busy everyday lives, the last thing many of us would think about is whether the cells in our bodies are dying the way they should.
But how and when our cells die is critical for our health and, when it goes wrong, it can contribute to cancer, infection, immune disorders and even our recovery from injury.
As humans, we have developed sophisticated mechanisms to ensure that when a cell is compromised – perhaps through bacterial or viral infection, or a genetic mishap –?it dies before it has the opportunity to impact our heath.
One of these ways is for the cell to sacrifice itself in a process termed ‘programmed cell death’. One well-studied mechanism of programmed cell death is called apoptosis, during which, the affected cell shrinks and collapses in on itself and is gobbled up and removed by immune cells silently, without a trace.
MLKL can be considered a zombie protein, because while it resembles an enzymatically-active protein termed a kinase, it is ‘catalytically-dead’.Cells can also die through injury. This might include physical trauma or chemical injury, such as alcohol exposure. This kind of cell death, called necrosis, is generally messy. The cell explodes and signals to the immune system that there is an issue resulting in inflammation. One example is liver necrosis, in which the liver becomes inflamed and partially dies due to prolonged excessive alcohol consumption.
In 2005, Degterev et al. first described a non-apoptotic form of programmed cell death that had the characteristics of necrosis. This new type of cell death was coined ‘necroptosis’ and opened up a new field in cell biology – to understand the signalling pathways that lead to necroptosis and the diseases consequences when necroptosis goes wrong.
Necroptosis is a type of programmed cell death that protects us against bacterial and viral infection by alerting the immune system of a potential attack. The critical difference to apoptosis is that the cell ‘sends’ a message into its local environment, recruiting the immune system to respond. The combination of cell death and immune response protects the organism from spreading viral or bacterial infections.
Targeting cell death to treat disease
We already know of a range of diseases associated with cell death disturbances.
Drugs that target apoptosis, such as BH3-mimetics, have been approved as anti-cancer treatments for some types of leukaemia, and are in clinical trials for other blood and breast cancers. More than 30 years of research into the fundamental biology of apoptosis, and a detailed understanding of the proteins that control cell death by apoptosis was key to the development of these drugs.
Dysfunctional necroptosis has been linked to a range of diseases, including cancers, autoimmune diseases, cardiovascular injury and stroke, and infection.
Switching off necroptosis to prevent inflammatory cell death could aid in the treatment of inflammatory diseases such as Crohn’s disease and inflammatory bowel disease and prevent brain injury after stroke. Alternatively, switching on necroptosis could be a way of treating cancers linked to a deficit in necroptosis, including breast, colorectal, melanoma and ovarian cancers.
The link between necroptosis and disease has created substantial interest in targeting necroptosis therapeutically. To do this well, it is essential to understand the proteins involved in the necroptosis pathways and how they interact with each other.
Killer zombie proteins
A significant amount of work over the past 13 years has begun to unravel the mysteries surrounding necroptosis. At the heart of the pathway sits the interaction between two proteins, RIPK1 and RIPK3, which form a still-poorly understood complex called the necrosome. In 2012, an unusual protein called MLKL was identified as the final essential player in the necroptosis signalling pathway.
MLKL can be considered a zombie protein, because while it resembles an enzymatically-active protein termed a kinase, it lacks key elements required to catalyse a chemical reaction and as such is ‘catalytically-dead’. While active kinases serve to pass a signalling baton between proteins in a pathway, the functions of zombie ‘pseudokinase’ proteins like MLKL have proven more elusive to grasp. In necroptosis, RIPK3 uses its enzymatic activity to mark MLKL and convert it into a killer protein. Because MLKL is so essential to necroptosis, when it is removed from cells in a laboratory, those cells can no longer die through necroptosis. It is also possible to genetically delete MLKL from mice and this creates a platform to study what happens in various diseases when necroptosis is switched off.
Since MLKL is expressed in most of our cells without killing them, we wanted to know more about what MLKL looks like in this dormant state and how it transitions into the killer protein.
In our recent work, published in Nature Communications, we used cutting-edge mass spectrometry to reveal that there is a two-step process for the MLKL protein to transition from its dormant to its killer form. It then self-assembles with three other MLKL proteins to form a complex made up of four MLKL proteins, or a tetramer, in order to kill the cell.
This finding was so significant because the killer assembly of mouse MLKL only contains three proteins (that is, a trimer) and was the first clue that the finer details of necroptosis is different in mouse and human cells.
MLKL in human cancers
Excess and uncontrolled necroptosis is associated with a number of inflammatory diseases. However more recently, we have begun to see that there is also a link between abnormal necroptosis and cancer development.
We see a number of cancers that have abnormalities in the MLKL protein. In order to test how these mutations affected necroptotic cell death in cancer scenarios, we generated human cells that recreated MLKL mutations found in colon, lung and endometrial cancers.
Cells expressing these MLKL mutations either took longer to die or did not die at all, because the mutation reduced or inhibited the ability of MLKL to form the killer assembly. Our task now is to work out if this delay or reduction in necroptosis caused by mutations in MLKL gives the tumours a growth advantage or makes them less responsive to current chemotherapies.
[caption id="attachment_69385" align="alignleft" width="581"]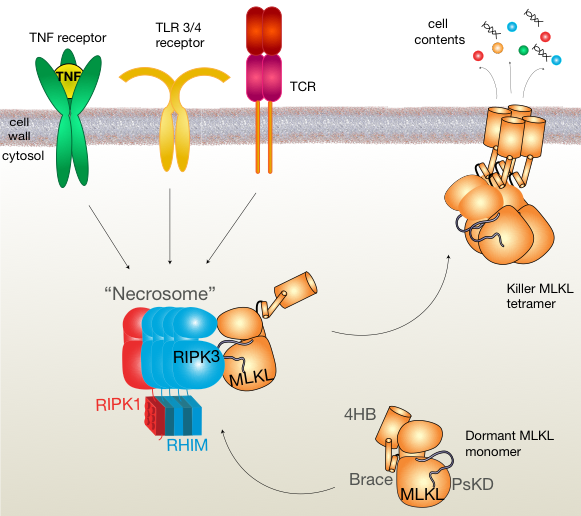 Figure 1: Signalling pathway that leads to necroptosis. Necroptosis can be activated downstream of the TNF, TLR3/4 and T-cell receptors and coalesce on the formation of RIPK1 and RIPK3 into a complex called the ‘necrosome’. MLKL is found in the cytosol on cells in a dormant state, and somehow is recruited to the necrosome and phosphorylated by RIPK3. This promotes the self-association of MLKL into tetramers in human cells that go on to kill the cell.[/caption]
Figure 1: Signalling pathway that leads to necroptosis. Necroptosis can be activated downstream of the TNF, TLR3/4 and T-cell receptors and coalesce on the formation of RIPK1 and RIPK3 into a complex called the ‘necrosome’. MLKL is found in the cytosol on cells in a dormant state, and somehow is recruited to the necrosome and phosphorylated by RIPK3. This promotes the self-association of MLKL into tetramers in human cells that go on to kill the cell.[/caption]
One of the most significant findings from our recent study is that the process of necroptosis has evolved in human cells to be much more regulated than in its mouse counterpart. In a laboratory setting, mutations can be made to the mouse MLKL protein that cause uncontrolled cell death, but equivalent mutations in human cells caused no cell death at all. This challenged our model system and suggested that the differences lie in the RIPK3:MLKL interaction, which we believe is more sustained in human cells.
The map to necroptosis is important for drug design
Search on the term ‘apoptosis signalling’ and you will find a number of very complicated pathways that intersect and interweave with each other. This fine level of detail is the result of decades of research in the apoptosis field, which remains a thriving research area. It is probably naïve then to expect that the necroptosis signalling cascade is as simple as RIPK1 activates RIPK3, which activates MLKL. However, it is clear these three proteins are certainly main players in the process and taking MLKL out of that pathway shuts down necroptosis.
It is no surprise then that many groups around the world are trying to work out how MLKL functions and forms that killer assembly, so that it can be controlled in different disease settings. In this context, our current work is very important as it highlights that the molecular details of MLKL killer assembly differs between organisms and this has implications for drug discovery.
Author:
[caption id="attachment_69386" align="alignnone" width="212"] Emma Petrie, Cell Signalling and Cell Death[/caption]
Emma Petrie, Cell Signalling and Cell Death[/caption]
Dr Emma Petrie is Senior Research Officer at the Cell Signalling and Cell Death Division of the Walter and Eliza Hall Institute of Medical Research in Australia
