A personal interpretation
25 Mar 2016 by Evoluted New Media
There is a clear move away from a ‘one size fits all’ approach to medicine and instead a new personalised medicine strategy is becoming more important. Mike Furness explains more about multi-omics.










There is a clear move away from a ‘one size fits all’ approach to medicine and instead a new personalised medicine strategy is becoming more important. Mike Furness explains more about multi-omics.
It has been known for a while that people with different genotypes respond to drugs differently. Knowledge gained from studying rare genetic disease has improved understanding of important biological pathways, creating the opportunity for more effective treatments.
Traditionally disease is defined by its symptoms and its location in the body.
For early developmental diseases this has meant that each symptom is investigated in isolation, by a specialist in that area. The patient is sent from one clinician to another. On average a child with a rare genetic disease will been seen by seven physicians over a five year period before a diagnosis may be found. For many of these children there will be no diagnosis but recent advances in genomics will address this problem.
It was against this background that the Discovering Development Disease (DDD) project was established between the NHS and the Wellcome Trust Sanger Institute. It has so far genotyped around 14,000 children with undiagnosed conditions and their parents, providing diagnoses for around 40% of these families, and identifying clusters of affected children that had similar clinical characteristics and shared damaging genetic variants in the same gene¹. Many of these genetic diseases are so rare that a clinician may see only one or two cases in a career; so being able to compare their patient’s genetics to this growing body of knowledge is a major step forward in helping consultants determine a definitive diagnosis.A system for annotating genes, developed initially from the Human Genome Project, was further developed to make it easier for non-specialists to see clearly which genes were associated with disease. Annotating the gene-disease associations will help others diagnose the disease in future. The study also found not all mutations were associated with disease. The knowledge emerging from this work has significantly advanced our understanding, not just the cause of disease but also in providing insights into important pathways controlled by these genes.
Technology developed by Wellcome Trust Sanger Institute scientists working on the DDD project supported the annotation and interpretation of the gene mutations, has been further developed by Congenica, a spinout from the Wellcome Trust Sanger Institute, to become Sapientia. This tool has now been validated by Genomics England and adopted as a genome interpretation platform for rare disease in the 100,000 Genomes Project. A number of genetics centres have worked closely with the development of this platform to ensure that it is designed to support clinical diagnosis. This includes work by Professor Phil Beales, Head of Genetics and Genomic Medicine at the Institute for Child Health at UCL, Director of the Centre for Translational Genomics and Head of the Cilia Disorders Laboratory (CDL). His team used Sapientia to provide a diagnosis of Bardet-Biedl Syndrome (BBS).
BBS is a genetic disease of the cilia, small rod-like structures that are found on the outside of almost all cells.
The physiological importance of cilia is still unclear, but they appear to be involved in various signalling pathways. There are several diseases relating to these structures but the diseases are difficult to diagnose, as they are present in almost all tissues so there is considerable variation in the symptoms. The main clinical features of BBS are obesity, rod–cone dystrophy, (causing night blindness followed by visual loss in childhood), specific learning difficulties in some but not all individuals, renal dysfunction and a range of secondary features such as extra fingers or toes. There is considerable variation in symptoms even within families.
In this case a family had two children with different symptoms, or phenotypes (Figure 1). Before arriving at Great Ormond Street they had tests at three different hospitals, including ECG, multiple blood tests and standard single or small number gene tests, which had not given any real answers. In addition, they had been seen by ophthalmologists, nephrologist, dieticians and psychologists.[caption id="attachment_52436" align="alignnone" width="620"]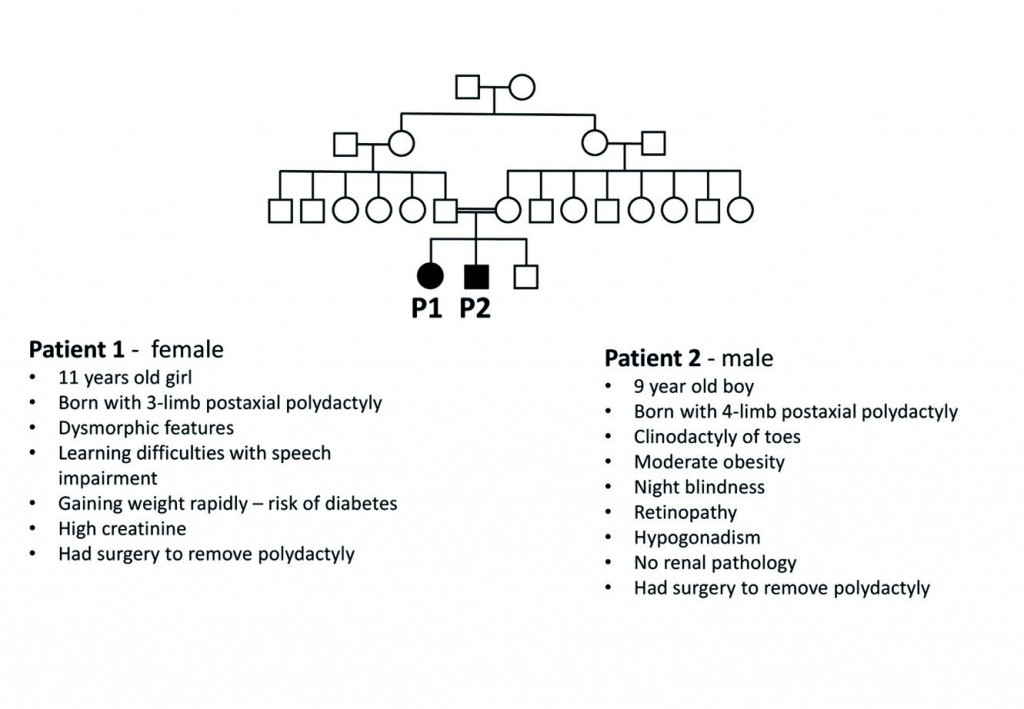 Consanguineous family (husband and wife are first cousins) with two affected siblings that have different clinical symptoms typical of BBS.[/caption]
Consanguineous family (husband and wife are first cousins) with two affected siblings that have different clinical symptoms typical of BBS.[/caption]
When they came to Professor Beales, an expert on ciliopathies, he sequenced the exome (the subset of the DNA that codes for protein) and then ran the results through Sapientia. This revealed that a whole section of the gene was missing in the patients and also discovered several other novel genes that appeared to be related to the patients’ disease. This knowledge allowed a more definitive diagnosis of BBS, identified more about the mechanisms of the disease and the genes that may be involved, and demonstrated that one factor was that the missing part of the genes for this protein led to a change in how it worked (Figure 2 and 3).
[caption id="attachment_52437" align="alignnone" width="620"]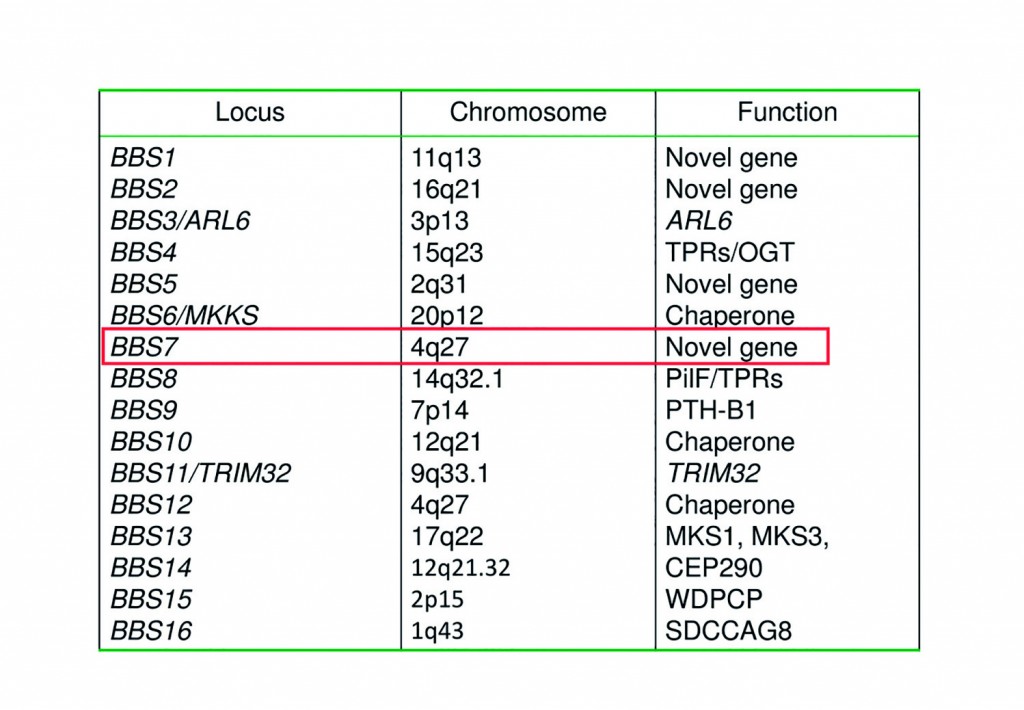 List of genes with potential mutations generated from current data analysis software. Further investigation is needed to see if BBS7's mutation is disease causing.[/caption]
List of genes with potential mutations generated from current data analysis software. Further investigation is needed to see if BBS7's mutation is disease causing.[/caption]
Unravelling how the mutation affects the metabolic pathways will improve treatments for rare diseases.
Known as orphan diseases, over 6,000 have been identified, of which only 600 have registries – lists of symptoms and other information associated with the disease. It is the patient support groups, helped by organisations such as EURORDIS (European Organisation for Rare Diseases) and the Genetic Alliance that are beginning to build upon the existing registries. This is marking a major change in the balance between the pharma companies and the patients.
[caption id="attachment_52438" align="alignnone" width="620"]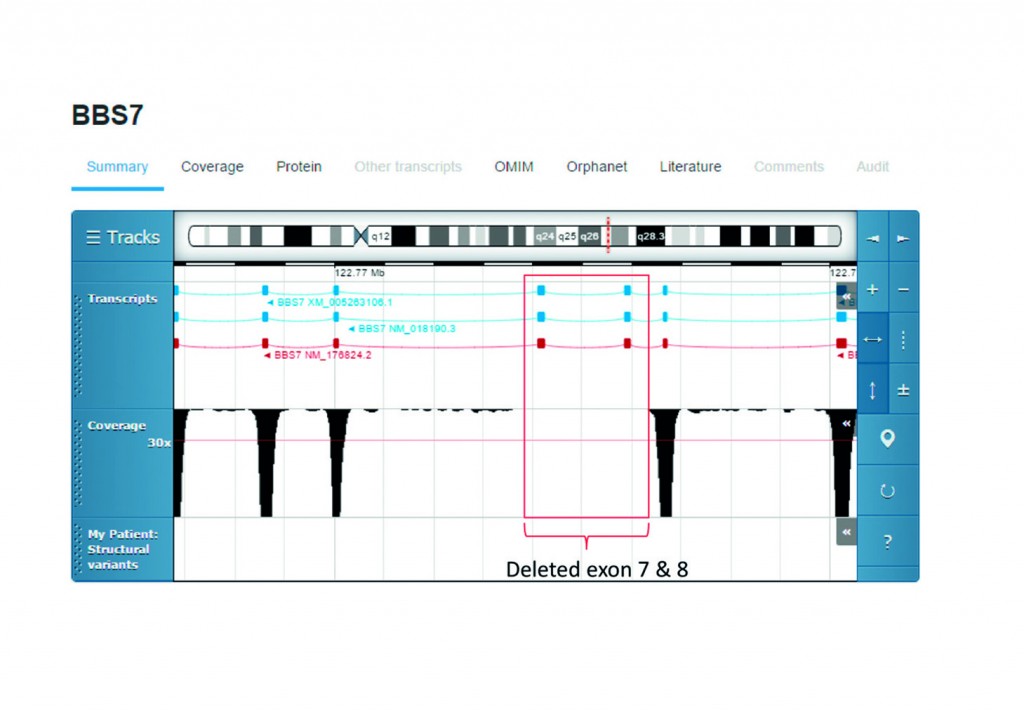 Visual representation of DNA sequencing data using Sapientia allows clinicians to see exon 7 and 8 are not present in the patient, when compared to a health sequence.[/caption]
Visual representation of DNA sequencing data using Sapientia allows clinicians to see exon 7 and 8 are not present in the patient, when compared to a health sequence.[/caption]
Many of these orphan diseases were previously considered to affect too few people to merit investment in a treatment. Now with improved knowledge of multi-omics, which combines information on the genome, the transcriptome (the RNA that codes the protein) and the proteome, (all the proteins), and other changes in the body, it is possible to understand in more detail the biology of the disorder or disease, and the mode of operation of existing drugs. This is enabling companies and support groups to identify those that could be repurposed for treatment for an orphan disease. The main cost of drug development is taking a drug through to Phase 2 testing, which can cost millions of pounds.
A deeper understanding of the mechanisms involved can increase the likelihood of success in Phase 2 trials.
Repurposing a drug for a new condition means taking a drug that has already passed all the safety and efficacy-testing run in Phase 1 and 2, and seeing how it can be used for a new condition. As it has passed on safety, the clinical trials can be started much more quickly and at much lower cost. Some of the larger patient groups are identifying drugs that are being used off-label for their condition and using social impact funding to drive small proof of concept studies. The rationale is that if the treatment is found to be effective they could go to the NHS and say “by using this treatment you will save £x” then ask for a percentage of that saving to reinvest in further research.
At the moment much of the work in rare genetic diseases has been directed at childhood developmental diseases but this is expanding to include late onset diseases. The big obstacle at the moment is that clinically you are not allowed to report on genes for which there is no therapeutic treatment or intervention that can ameliorate the condition. In the United States, there is a list of around 50 of the 22,000 known genes that can be reported on, as these are clinically actionable, i.e. there is a form of intervention known to be beneficial.
In research there is not the same limitation; the approach is “we don’t know what is causing this disease, so let’s look at everything”.
So researchers look at all the genes that have mutations, the differential expression levels in normal and diseased tissues, and the differences in proteins and metabolites. By identifying which of these are altered in the disease, we can begin to build an understanding of the underlying biology. Pharmacogenomics started in the 1960s when an enzyme – cytochrome p450 2D6 – was discovered to be important for drug metabolism. Since then, around 300-400 other genes have been shown to play significant roles in how drugs are metabolised in the body. There can be considerable variation between individuals in regards to the efficiency of the drug and amount of the proteins made by these genes. Having multiple copies of the relevant gene means that certain drugs are broken down quickly, reducing the time the drug can provide beneficial effects, whilst reduced or no copies meant drugs stay in the body longer, potentially increasing the likelihood of toxicity.
Among these genes are a family called the HLA genes. It was found that HLA profiles of individuals could provide a good indication of how well a transplanted organ will be received so individuals are matched with donors to avoid rejection by the recipient’s immune system. So before an organ transplant patients are both blood and HLA typed. The problem with these genes, and many others, is their length. For example, in Huntington’s Disease, patient’s genes contain many repeated sequences, with the amount of repetition being linked to the disease. However, sequencing them reliably, knowing how many of the repeats are genuine and how many are artefacts, is very difficult using the sequencing technology currently used today.The original Sanger sequencing technology generated reads that were several hundred bases long, compared to the current predominant technology using Illumina short reads only a couple of hundred bases in length. Long read technologies can now generate reads of several thousand bases, but can cost 10-20 times as much as Illumina’s $1,000 per genome. However, increasing the read length in sequencing technology allows more accurate identification of large structural changes in the genome, which cannot easily be identified by short reads. The current consensus is to do a small amount of long reads and effectively use that as a framework on which to build the short reads, improving accuracy at a reduced cost. It is likely that we will see many developments in this area but all the sequencing technologies are working within an established schema and produce a VCF file, (a standard format for sharing information) which means that technologies downstream such as Sapientia are future proofed to these developments.
[caption id="attachment_52456" align="alignnone" width="540"] The quicker a patients DNA sequence can be looked at and interpreted, the quicker they can receive medical assistance.[/caption]
The quicker a patients DNA sequence can be looked at and interpreted, the quicker they can receive medical assistance.[/caption]
Speed of diagnosis allows the most appropriate drug to be administered quickly. For example, patients diagnosed with breast cancer are currently genotyped to identify which drugs they are most likely to respond to, speeding up patient recovery whilst also reducing the costs spent on ineffective drugs.
Another development that will allow genotyping to be more readily available is the emerging knowledge that only a small part of the exome is linked to disease.
A paper by the Wellcome Trust Sanger Institute studied the genomes of people from the 1000 genomes pilot project people without disease. It found that there were around 400 damaging variants in each person and 2 bona fide disease variants². In addition, recent evidence from the MedSeq project at Brigham & Women’s Hospital³ looked at 4,600 disease-related genes in 100 healthy individuals and showed that 92% of the people were carriers for at least 1 disease variant, and 21% showed variants for dominant Mendelian conditions.
Traditionally, the process of finding a mutation that caused a disease was a manual operation that could take three to four weeks for each patient. It would produce a list of variants several pages long and someone would have to search through the literature for information on the genes, cross-referencing them with the biological pathways thought to be involved, to slowly build up an understanding of what was happening. Now with Sapientia that process is automated. Taking the genetic and phenotype information, it highlights the mutations that may be linked to disease from the sequencing data and links that to databases of knowledge about previously seen variants, phenotype information and a wide range of literature sources. As more of this data is added to the knowledge base, the accuracy in identifying the critical sequences increases.Over the last 12 months Sapientia has been rolled out across the NHS, over half the Regional Genetics Centres are now introducing the technology and migrating to the new platform.
This is bringing us to a situation where a patient could be profiled and matched with the most appropriate therapies and also advised on any potential adverse interactions with other drugs such as painkillers, based on their genetics. You can imagine a time in the near future where the patient is tested and within a few days they can be told which significant genetic variants their genes contain, and as a result the treatment options available. The conversation may go “having checked your drug metabolising enzymes two of these drugs may not be suitable, so we’ve selected this one that will give you the best chance of positive outcome”.
NHS England announced its Personalised Medicine Strategy in September 2015? , this sets out how emergent approaches in areas such as diagnostic testing, functional genomic technologies, molecular pathways and data analytics will be harnessed to better manage patient health and target therapies. It is clear that emerging knowledge of multi-omics, and tools such as Sapientia, are enabling this knowledge to be captured and accessed by clinicians across the NHS, helping to embed genomic technologies into clinical pathways, and supporting the NHS in becoming one of the most advanced healthcare systems in the world.Author
Mike Furness of Wellcome Trust Sanger Institute spinout Congenica
References1. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet (London, England) 2015;385;9975;1305-14 PUBMED: 25529582; PMC: 4392068; DOI: 10.1016/S0140-6736(14)61705-0 2. Deleterious- and Disease-Allele Prevalence in Healthy Individuals: Insights from Current Predictions, Mutation Databases, and Population-Scale Resequencing Am J Hum Genet. 2012 Dec 7; 91(6): 1022–1032. doi: 10.1016/j.ajhg.2012.10.015 3. Highlights from the MedSeq Project: ClinGen Meeting May 27, 2015 Robert C. Green, MD, MPH director, genomes2people Research Program in Translational Genomics and Health Outcomes Division of Genetics, Department of Medicine, Brigham and Women’s Hospital 4. Available online.
